特長
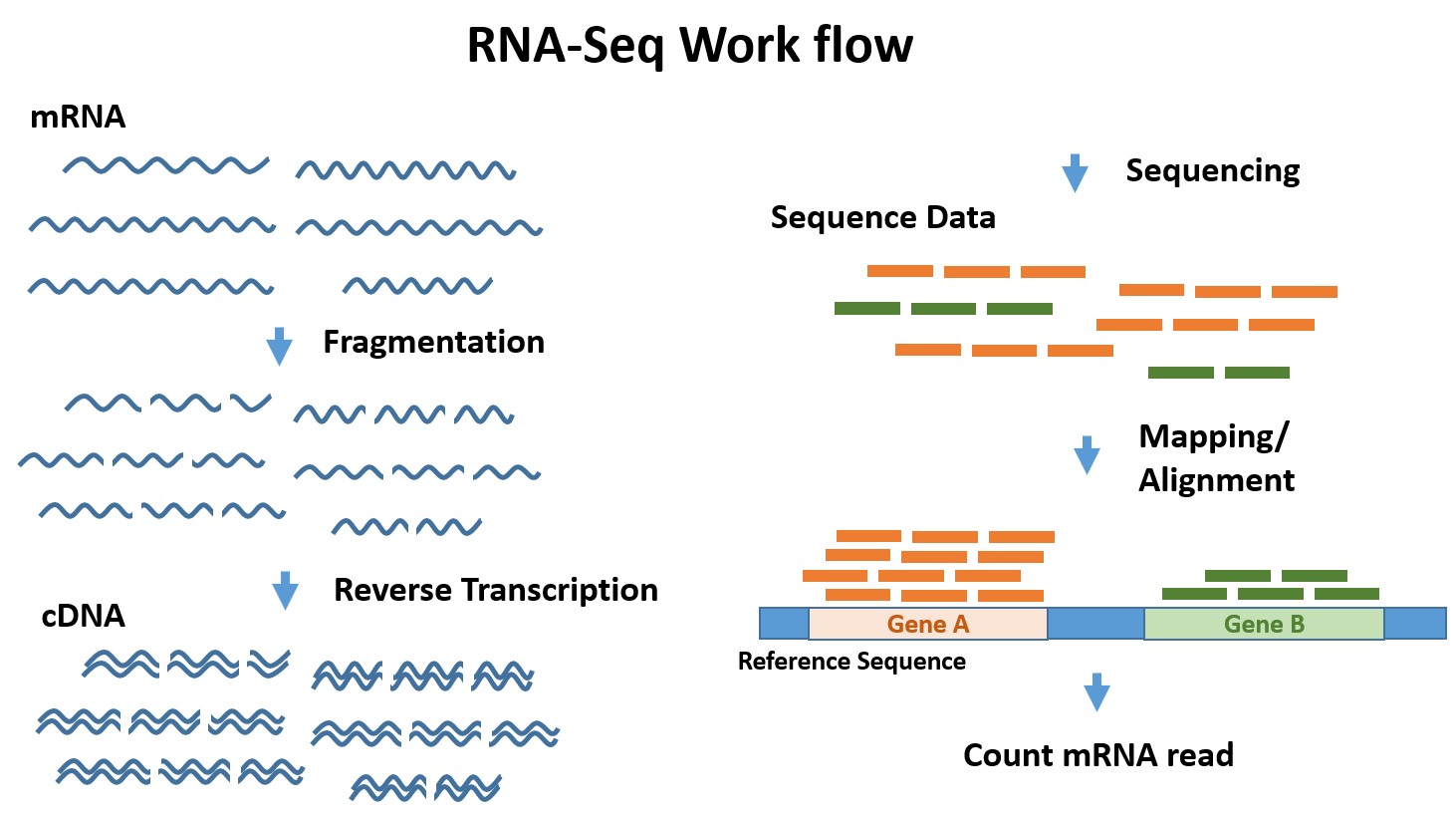
短いリード長(50~100bp)に断片化されたcDNAを超並列で高速解析することで、トランスクリプトーム(転写物)を網羅的に取得し、遺伝子発現レベルを定量します。次世代シーケンサーを用いたRNAシーケンスでは、既知の遺伝子発現解析だけでなく、マイクロアレイでは解析できない新規転写産物の検出やスプライシングバリアントの検出、融合遺伝子の検出が可能です。またシーケンス量を大きくすることで、より低発現の転写産物の検出が可能になります。
取得データ量は4Gbから承ります。ご希望のシーケンス深度(sequencing depth)をお申し付けください。
クラボウのRNA-Seq解析サービスでは、「ライブラリ調整~シーケンス(FASTQデータ)」と「データ解析」サービスをご用意しています。
解析方法
Total RNAからpolyA RNAを精製、逆転写反応で合成したcDNAをシーケンシングします。
ヒト・マウス・ラットなど哺乳動物を対象にした遺伝子発現解析の場合、シーケンス方法は100bpペアエンドシーケンス、取得リード数は40million readでの解析を推奨しています。
データ解析においては、既知のゲノム配列にマッピングし、遺伝子領域内にマッピングされたリード数をカウントし、遺伝子領域長で補正されたデータ(FPKM値)を遺伝子発現値として算出します。
●標準解析内容
・使用キット: TruSeq Stranded mRNA Library Prep Kit (illumina社)
・ライブラリータイプ: Paired-end ライブラリー
・シーケンサー: illumina NovaSeq6000
・リード長: 100bp
必要サンプル条件
Total RNA 3.0μg以上(濃度 100ng/μL以上)
RIN 7以上、rRNAピーク比 1以上
*品質 RIN 7以上、rRNAピーク比 1以上
*サンプルはヌクレアーゼフリー水に溶解してください
*RNAの定量はQubit Fluorometerなどの蛍光法による測定データをご利用ください。NanoDropなどの吸光度による測定データは過大評価される恐れがあります。
解析結果
データ量により、DVD-R、USBメモリ、HDDのいずれかで納品します。
●シーケンス解析のみをご依頼の場合
・解析報告書(シーケンス結果サマリー):解読された塩基数やクオリティスコアなどを含みます。
・サンプル初期チェック結果報告書
・シーケンスデータ一式(Fastq形式)
遺伝子発現解析(:Expression profile)をご依頼の場合【有償オプション】
・トリミングおよびマッピング結果のサマリー(解析報告書に追加)
・RAWデータの品質チェック結果(html形式)
・遺伝子発現解析結果(Excel形式): 対象のゲノム領域にマッピングされたRead count、FPKMおよびTPM値を報告。
詳細は次の通りです。
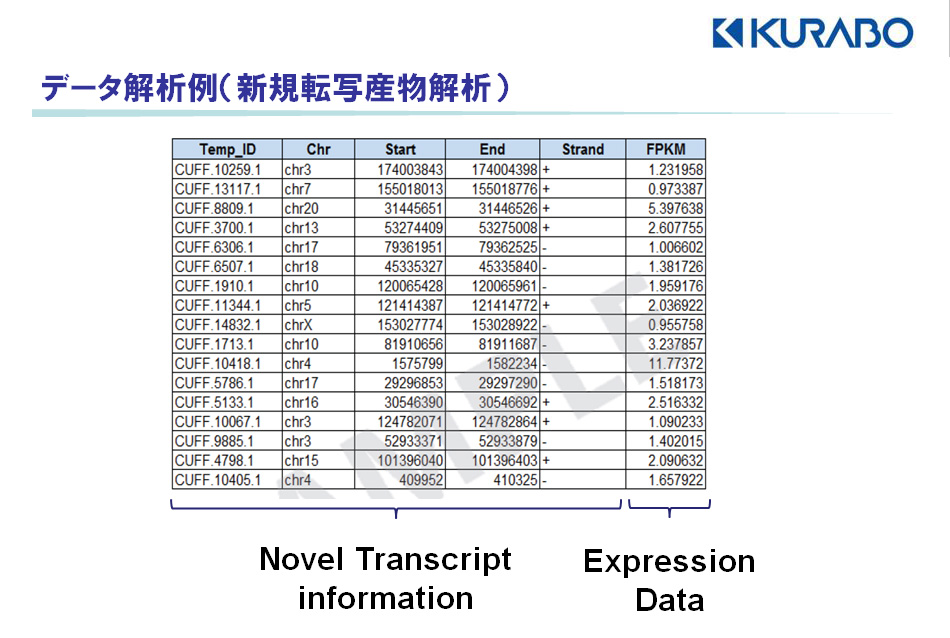
【新規転写産物の検出】
マッピングされた染色体の位置情報(染色体番号、塩基番号)、遺伝子コード方向(フォワード鎖/リバース鎖)、発現値(FPKM値)をエクセルファイルにて報告します。(
fig.1)
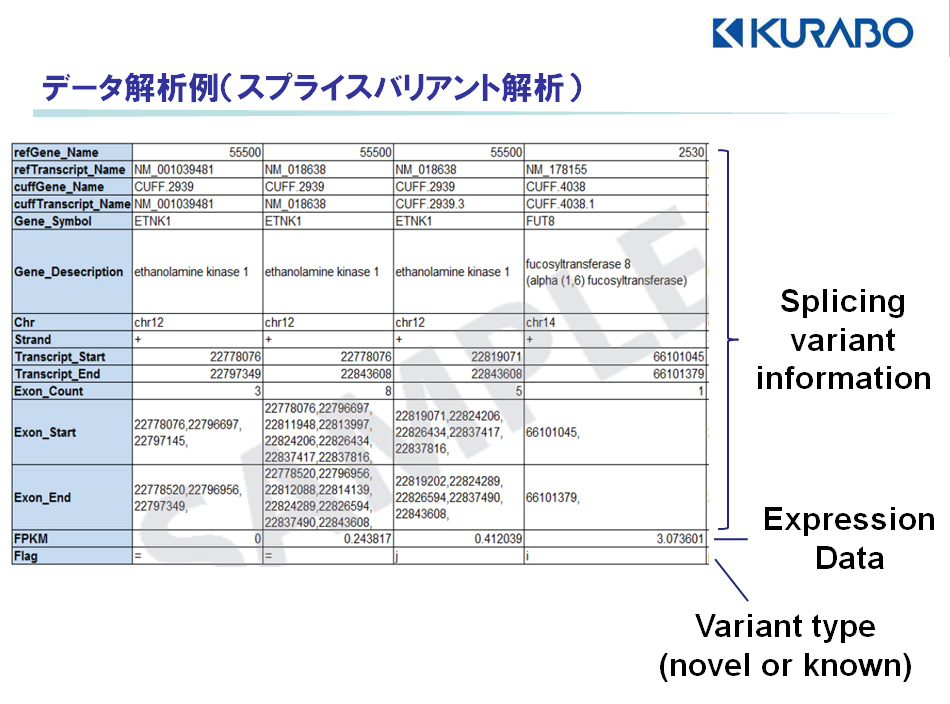
【スプライシングバリアントの検出】
バリアントを検出した遺伝子の情報(遺伝子名、データベースID、染色体位置)、バリアントに関する情報(エクソン数、エクソン開始・終止場所)、発現値(FPKM値)をエクセルファイルにて報告します。(
fig.2)
【融合遺伝子の検出(ヒトのみ)】
検出した融合遺伝子の情報(塩基配列数、塩基配列情報)、由来となる各遺伝子の情報(遺伝子名、データベースID、染色体位置、遺伝子コード方向、染色体開裂場所)、融合遺伝子が起こっている確率をエクセルファイルにて報告します。(
fig.3)
* シーケンス解析のみをご依頼の場合には、FASTQファイルでの納品となります。上記はデータ解析を依頼いただいた場合の結果例です。
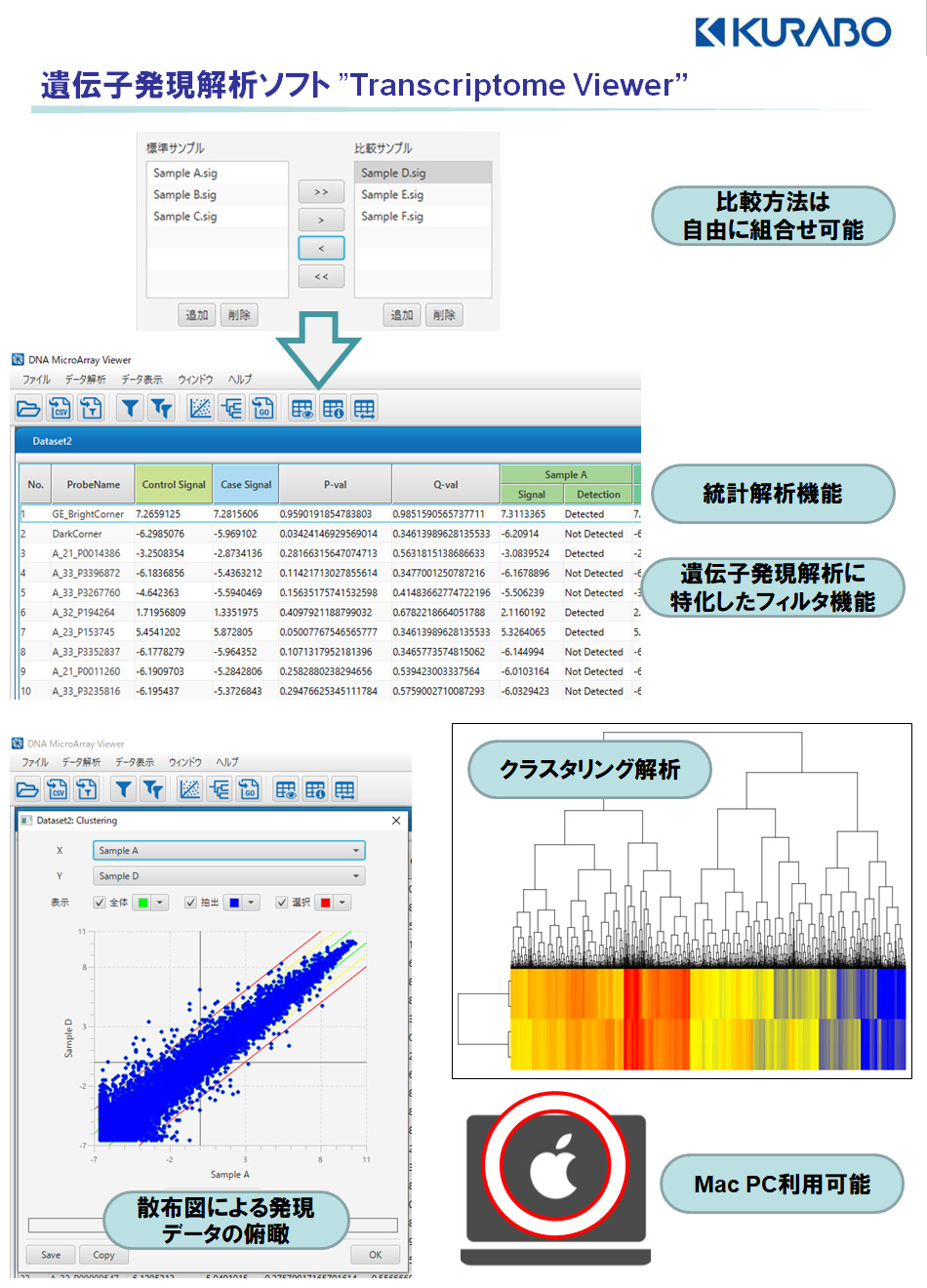
発現比較解析(:Differential Expression Gene)をご依頼の場合【有償オプション】
データのご指定のない場合、遺伝子発現解析(Expression profile)で算出したread count値で解析します。また抽出条件のご指定がない場合、クオリティの低いデータをフィルターし、|Fold Change|>=2で抽出します。* パラメータや報告内容の変更をご希望の際はご相談ください。
・比較解析結果レポート(html形式)
-比較解析に使用するデータの品質チェック結果(histogram)
-サンプル間の比較・相関分析(box plot, Density plot, Correlation matrix, MDS (Multidimensional Scaling) plotなど)
・クラスター解析結果(png形式)
GO解析(:Gene Ontology)をご依頼の場合【有償オプション】
g:Profiler toolを使用して、GOエンリッチメント解析を実施します。 各遺伝子には詳細な情報(アノテーション)が付与されていますが、その中でもGene Ontology(GO)は貴重な情報を入手することができます。 GOは「cytokine signaling」「apoptosis」「cell differentiation」など、遺伝子が持つ機能を体系的にまとめたデータベースです。 各遺伝子には複数のGOが紐づけられており、聞きなじみのない遺伝子がリストアップされたとしても、これらの情報から遺伝子の機能を類推することができます。
・Dot plot、histogram(各カテゴリの上位のGO termを示す)(png形式)
・抽出遺伝子リスト(Excel形式)