特長
mRNA-Seq 遺伝子発現量解析[mRNA-Seq 遺伝子発現量×NovaSeq バリュープライス]
遺伝子配列リソースが充実した生物種における、mRNA-Seqによるサンプル間の遺伝子発現量(Differential Expression)解析を行います。バイオインフォマティックスでは、マッピング、遺伝子発現量解析をオプション解析することができます。
- mRNA精製:ポリA精製 or rRNA枯渇
- Strand specific ライブラリ調製
- バイオインフォマティクス解析:マッピング、遺伝子発現量解析
サービス内容
- 受領したRNAサンプルについて、品質確認(サンプルQC)を行います。
- 目的に応じたRNAの精製を行います。
ポリA精製 (*1)もしくは、rRNA枯渇処理(*2)
- 精製したRNAの断片化を行います。
- ランダムプライマーを用いた逆転写反応により、cDNAを合成します。
- アダプター付加、2nd Strand cDNAの断片化を行い、Strand-specific mRNAライブラリーを調製します。
- 調製したライブラリーをシーケンシングします。
- (オプション解析)取得した配列を参照配列(*3)にマッピングし、遺伝子発現量の定量解析などを行います。
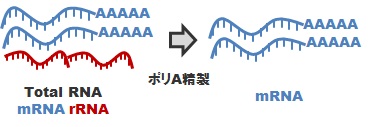
*1 mRNA-Seqライブラリ調製:ポリA精製(fig.1)
真核生物を対象とした標準ライブラリ調製方法です。total RNAをサンプルとしてお預かりして、ポリA精製を行い、mRNAを精製します。このmRNAをcDNA化し、所定のアダプタを付加することにより、mRNA-Seq(Strand Specific)ライブラリを調製します。
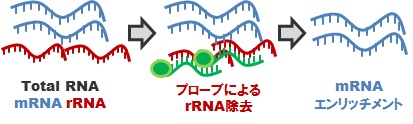
*2 mRNA-Seqライブラリ:細菌の場合(fig.2)
rRNA 枯渇処理 (通常はIllumina RiboZeroを使用)
原核生物(細菌)の場合、mRNA は原則として ポリA構造を持たないため、これを用いた mRNA 精製が困難です。かといって精製を行わないでシーケンシングをしても、rRNA配列ばかりを読んでしまうことになります。細菌トランスクリプトーム解析を実現するためには、予めプローブによる rRNA枯渇処理を行ってから、cDNA化、ライブラリ調製を行う必要があります。生物種により相性がありますので、予めご相談ください。なお、実際のrRNA枯渇の程度については、保証することはできません。
ヒト・マウス・ラットのtotal RNAサンプルからも、rRNAを効率よく除去することができます。断片化が進んだFFPEサンプルのrRNA除去に有効なだけではなく、ポリA精製によって失われていた、ポリA末端を持たないトランスクリプトをシーケンス解析対象とすることが可能となります。詳細については、お問い合わせください。
*3 参照配列
標準サービスでは、mRNA配列を参照配列としたマッピングを行います。
オプション解析として、参照ゲノム配列を対象としたマッピングを行います。