概要
タンパク質の機能は、その構造によって決まります。タンパク質が他のタンパク質、薬物、小型リガンド、または化学的誘導体と相互作用して、その構造をどの様に変化させるかを理解することは、生物学的プロセスを理解するために不可欠です。今日まで、タンパク質構造および立体配座変化を知るために、様々な技術が開発されてきました。しかし、それらのほとんどは、単離、精製されたタンパク質にのみ適用可能であるか、タンパク質もしくは低分子のいずれかを標識する必要があり、標的に対して高い親和性を持つ低分子にのみ適しているものばかりでした。
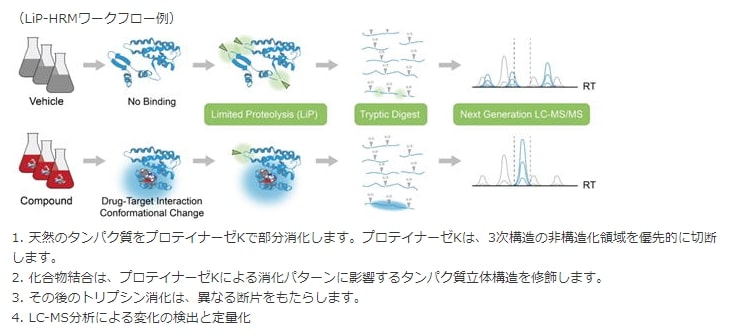
限定分解(Limited Proteolysis:LiP)は、未精製溶解物中のタンパク質構造の変化を同定および分析するためのラベルフリー技術として開発されました。LiPの基本概念は、化合物の存在下または非存在下で分解を受けるタンパク質に基づきます。化合物がタンパク質に結合すると構造変化を起こし、結合部位はタンパク質分解にアクセスできない状態になります。これらの効果がタンパク質分解のパターンを変化させ、質量分析によって特徴付けられるユニークなペプチド形成をもたらします。
得られたペプチドは、Biognosys社の独自技術であるHRMを使用して定量化されます。包括的なHRMデジタルマップによって検出可能なすべてのペプチドシグナルを取得できるため、LiPとHRMの組み合わせは、タンパク質の構造変化のプロファイリングに特に適しています。
特長
質量分析と組み合わせた限定分解(LiP-MS)は、化合物結合により誘導されたタンパク質構造変化を検出します
チューリッヒ工科大学のDr.Paola Picotti教授の研究室は、プロテオームワイドに化合物との相互作用によって引き起こされるタンパク質の立体構造変化に対処することができる、質量分析と組み合わせた独自のLiPプロトコル(LiP-MS)を開発しました。
このLiP-MS技術は、二重プロテアーゼ消化に基づいています。天然条件下での最初のプロテアーゼ工程は、タンパク質中に部位特異的切断を生じ、それらは切断時のタンパク質の立体構造に特異的であるため、いわゆる「同型」ペプチドをもたらします。非天然条件下での第二のプロテアーゼは、完全なタンパク質消化をもたらし、ボトムアッププロテオミクスによるペプチドの同定および定量化を可能にします。
より複雑な細胞環境におけるターゲットデコンボリューション
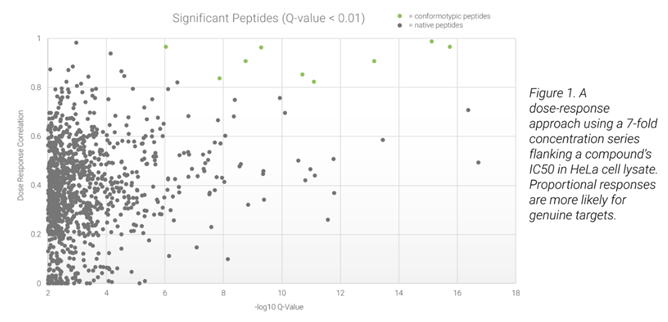
LiP-MS技術は、大規模なプロテオーム解析において、酵母および細菌において有望な結果を示しています。Biognosys社は、ヒトなどのより複雑な生物の分析のためにこの研究を拡大しました。生物のプロテオームがより複雑になるにつれて、化合物と相互作用する可能性のある潜在的な標的タンパク質の数が増加し、より多くの「ノイズ」につながります。この分析上の課題を克服するために、Biognosys社は用量反応アプローチを導入しました。タンパク質の立体構造変化と化合物の濃度との間の相関関係を示すことによって、シグナルノイズ比を高めることができました。これにより、複雑なプロテオーム環境下においても、低分子-タンパク質間相互作用の信頼性の高い予測が可能になります。
また、有意なペプチドが用量反応および高いq値に対する正の相関を介して同定されます。(
fig.2)
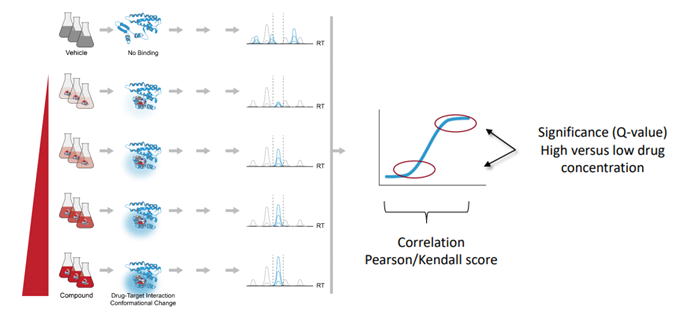
用量反応曲線を用いたLiP候補の検出
用量反応アプローチでは、いくつかの化合物濃度パターンから相関カーブを作成し、各ペプチドのLiPスコアを計算します。(
fig.3)
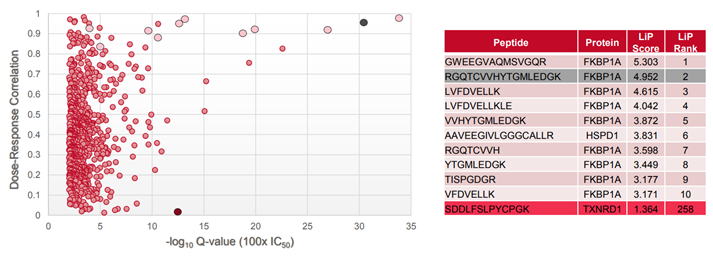
計算された総LiPスコアから最も可能性の高い標的タンパク質を同定します。(
fig.4)は、ラパマイシンで処理されたHeLa細胞を解析した場合の用量反応相関図とLiPスコアテーブルです。LiPスコアが高い上位10ペプチドのうちの9ペプチドは、ラパマイシンの既知の標的タンパク質(FKBP1A)と対応しました。
【プロジェクト例】
- 細胞数:5x107~1x108個の凍結ペレット
- 化合物条件:最大投与量で20μL(IC50の10,000倍)
- サンプル条件:解析サンプル7用量点、DMSOコントロール×1、ポジティブコントロール×1(各N=4、合計36サンプル)